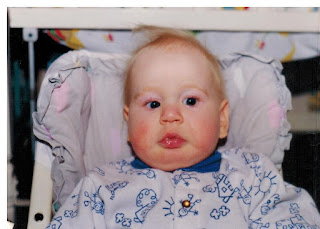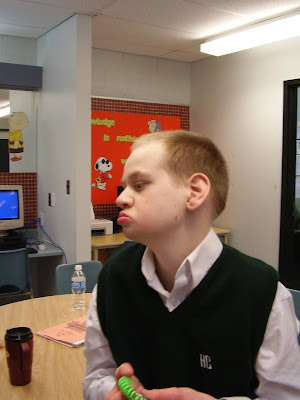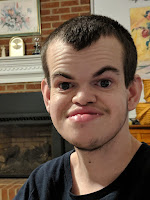
We were living in Saskatoon, Canada. It was Good Friday, April 9th, 1993 when our Atle decided to come into this world. He was such an easy delivery and everything seemed fine. He looked perfect. No obvious abnormalities, no problems initially. I was a bit worried because he didn’t latch on well to the breast but his older brother also took a while to get the hang of it so it wasn’t a big concern. They told me that his blood sugar was low the next morning so we stayed an extra day. But he still didn’t seem to be able to latch. He would paste his tongue up to the roof of his mouth leaving no room for the nipple. My brilliant husband figured out how to use one of those pacifiers with the hole in the back for your finger to force the tongue down so that we could slip the nipple in and get him to eat. Breastfeeding was not going to happen for this one apparently. So, we went home with the formula a bit disappointed but no big deal.
At home we noticed that he ate very very slowly. Like over an hour for one of those mini 4-ounce bottles. He would completely exhaust himself with eating and fall asleep before he could finish. The next day I couldn’t wake him up at all. I undressed him, used a wet washcloth, even pinched him but couldn’t wake him up for more than a few minutes. Off to our trusted family doctor. There we discovered a huge inguinal hernia protruding through his abdominal wall. He was 10 days old and already having his first surgery. The other side popped out a few days later and his second surgery was at 30 days of age.
So, when he still didn’t have head control and my MD brother-in-law was getting concerned, we just thought “well, he’s had two surgeries, he’ll catch up”. During this time, we began to notice breathing problems. The first time he had pneumonia, he tested positive for RSV. A lot of kids get that so nothing unusual there. But then he got pneumonia again and again and he still didn’t have good head control so the hospital staff became concerned and ordered a head CT. I will never forget a young neurologist who informed us that our child had global brain atrophy and in a moment of spectacularly poor bedside manner asked us if we knew what “retarded” meant and, “Oh, by the way, we need the bed. You are discharged and have 20 minutes to clear the room.”
We had been told that these things just happen and we would likely never find out a reason. But a wonderful geneticist ran tests and we discovered that our child had partial trisomy 11:22 (as it was known at that time). My husband was determined to be the carrier and after speaking with family members and their test results, we were able to trace the carriers back five generations.
We started physical therapy, but Atle also was still eating very slowly, having frequent pneumonia and breathing difficulties. He was being hospitalized at least one week out of every month including a life-threatening bout with tracheitis. He was tested several times for aspiration during this period but the test was always negative.
A job opportunity took us from our home in Canada to Wisconsin when Atle was three. Our first week in our new home was quite eventful. Atle had his first seizure while in the bath and stopped breathing. Resuscitating your own child is something I pray you never need to do. He was flown by helicopter to UW Madison Children’s Hospital. I am still thankful for their wonderful and thorough staff. More tests were run and the aspiration was finally proven. Atle received his g-tube on his 5th birthday. He was 25 lbs. I remember the look of relief on his face when he was tube fed for the first time. Finally, he could be satisfied without pain. He had been hospitalized over 70 times at this point with pneumonia. Unfortunately, he also aspirates his own saliva so he is still at great risk.
At this point, we moved down to North Carolina. There have been many more hospitalizations since. It was discovered that he only has one kidney and as he grew, the kidney has not kept up so he is in stage 3 renal failure. The renal failure affected his bone density so he broke his arm, his ankle and he also has scoliosis and kyphosis. He was having reflux aspiration resulting in Nissen fundoplication surgery. He had malrotation of the gut that was repaired surgically. As well as numerous ear tube surgeries and a severe fungal infection of the ears.
Atle is currently 28 years old and has a comprehensive routine to maintain his lung and renal health. He uses a therapy vest to loosen mucous twice daily, four nebulizers twice daily, medications for blood pressure, seizures, prophylactic antibiotics, probiotics, hypothyroidism, hyperparathyroidism, calcium, vitamin D and we also carefully monitor his fluid intake. All of these treatments have been paying off. Up until a recent hospitalization for Covid in February he was two years hospital free!
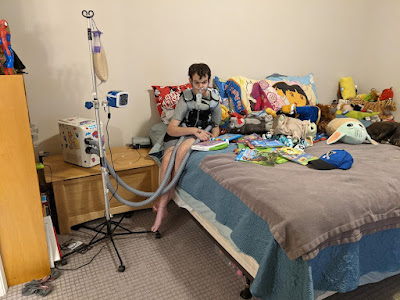
But that is just half the story. The real story of Atle is how smart and fun he is. Atle walks but does not talk. He uses signs, an iPad and gestures to communicate and is remarkably talented and making sure he gets his point across. He loves animals and knows all of their ASL signs. He plays Miracle League baseball, rides horses and plays sled hockey. He can do puzzles, dribble a basketball and help with chores. He loves music and leads the clapping in church. He is basically famous in our town as I cannot take him anywhere without people knowing his name and saying hello!
At home we noticed that he ate very very slowly. Like over an hour for one of those mini 4-ounce bottles. He would completely exhaust himself with eating and fall asleep before he could finish. The next day I couldn’t wake him up at all. I undressed him, used a wet washcloth, even pinched him but couldn’t wake him up for more than a few minutes. Off to our trusted family doctor. There we discovered a huge inguinal hernia protruding through his abdominal wall. He was 10 days old and already having his first surgery. The other side popped out a few days later and his second surgery was at 30 days of age.
So, when he still didn’t have head control and my MD brother-in-law was getting concerned, we just thought “well, he’s had two surgeries, he’ll catch up”. During this time, we began to notice breathing problems. The first time he had pneumonia, he tested positive for RSV. A lot of kids get that so nothing unusual there. But then he got pneumonia again and again and he still didn’t have good head control so the hospital staff became concerned and ordered a head CT. I will never forget a young neurologist who informed us that our child had global brain atrophy and in a moment of spectacularly poor bedside manner asked us if we knew what “retarded” meant and, “Oh, by the way, we need the bed. You are discharged and have 20 minutes to clear the room.”
We had been told that these things just happen and we would likely never find out a reason. But a wonderful geneticist ran tests and we discovered that our child had partial trisomy 11:22 (as it was known at that time). My husband was determined to be the carrier and after speaking with family members and their test results, we were able to trace the carriers back five generations.
We started physical therapy, but Atle also was still eating very slowly, having frequent pneumonia and breathing difficulties. He was being hospitalized at least one week out of every month including a life-threatening bout with tracheitis. He was tested several times for aspiration during this period but the test was always negative.
A job opportunity took us from our home in Canada to Wisconsin when Atle was three. Our first week in our new home was quite eventful. Atle had his first seizure while in the bath and stopped breathing. Resuscitating your own child is something I pray you never need to do. He was flown by helicopter to UW Madison Children’s Hospital. I am still thankful for their wonderful and thorough staff. More tests were run and the aspiration was finally proven. Atle received his g-tube on his 5th birthday. He was 25 lbs. I remember the look of relief on his face when he was tube fed for the first time. Finally, he could be satisfied without pain. He had been hospitalized over 70 times at this point with pneumonia. Unfortunately, he also aspirates his own saliva so he is still at great risk.
At this point, we moved down to North Carolina. There have been many more hospitalizations since. It was discovered that he only has one kidney and as he grew, the kidney has not kept up so he is in stage 3 renal failure. The renal failure affected his bone density so he broke his arm, his ankle and he also has scoliosis and kyphosis. He was having reflux aspiration resulting in Nissen fundoplication surgery. He had malrotation of the gut that was repaired surgically. As well as numerous ear tube surgeries and a severe fungal infection of the ears.
But that is just half the story. The real story of Atle is how smart and fun he is. Atle walks but does not talk. He uses signs, an iPad and gestures to communicate and is remarkably talented and making sure he gets his point across. He loves animals and knows all of their ASL signs. He plays Miracle League baseball, rides horses and plays sled hockey. He can do puzzles, dribble a basketball and help with chores. He loves music and leads the clapping in church. He is basically famous in our town as I cannot take him anywhere without people knowing his name and saying hello!